Method¶
FANTASIA is a modular pipeline for protein function annotation based on deep learning embeddings. Inspired by the GOPredSim algorithm [1], it performs large-scale GO term transfer by comparing learned vector representations of proteins, rather than relying on sequence alignment or homology alone.
The method is organized into three main stages:
Embedding Computation: Input protein sequences are transformed into high-dimensional vectors using pretrained protein language models (PLMs). Each model captures distinct biochemical and structural properties, enabling generalization beyond traditional sequence similarity.
Similarity Search and Annotation Transfer: The computed embeddings are compared to a reference database of experimentally annotated proteins. Functional annotations (GO terms) are transferred from the most similar entries, based on a learned distance metric. Optional redundancy and taxonomy filters can be applied to fine-tune the reference space.
Post-processing and Filtering: FANTASIA assigns a Reliability Index to each prediction, collapses redundant GO terms based on ontology structure, and optionally computes pairwise global alignments to assess sequence-level similarity. The results are then filtered, deduplicated, and structured for downstream analysis.
The pipeline is designed to be reproducible, extensible, and interpretable, suitable for both exploratory use and high-throughput functional annotation projects.
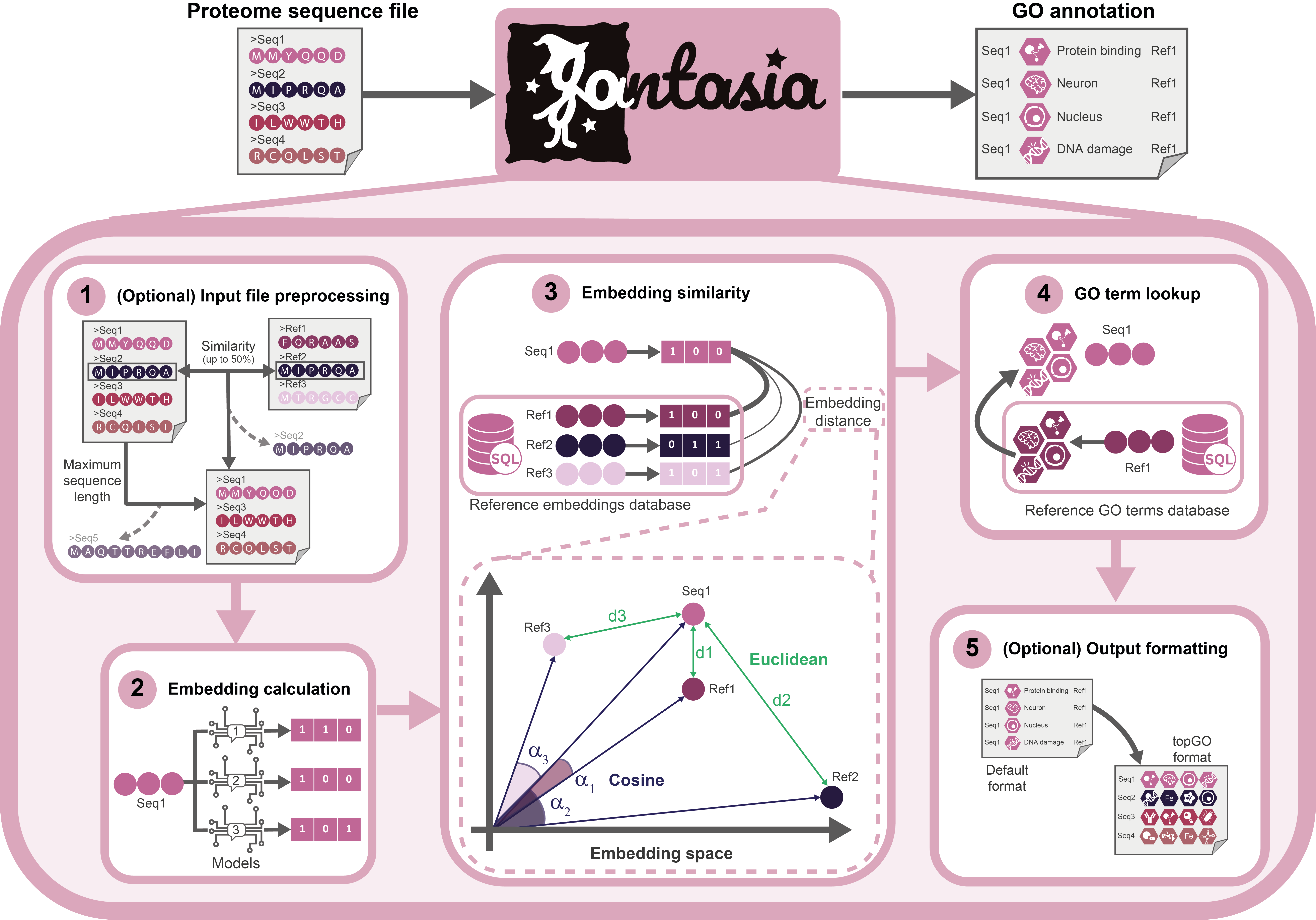
Step 1: Setup and Input File Preprocessing¶
Before computing embeddings, FANTASIA prepares the working environment and validates the input data. This step ensures that all dependencies are active, configuration parameters are correctly parsed, and the experiment directory is ready to store outputs.
The pipeline accepts a FASTA-formatted file containing one or more amino acid sequences. These sequences are parsed using BioPython and can be optionally filtered by length, according to the configuration. The input file is then split into batches that are distributed across the enabled protein language models (PLMs), such as ProtT5 [3], ESM2 [4], or ProstT5 [5].
Internally, this step performs the following actions:
Loads and merges the base configuration file (.yaml) with any overrides passed via command-line arguments.
Creates a timestamped directory for the current experiment, where all intermediate and final outputs will be stored.
Initializes the embedding environment by loading the selected PLMs and their tokenizers dynamically.
Parses and batches the input sequences, applying optional filters such as maximum length or execution limits.
Once the setup is complete and the sequence batches are prepared, the pipeline proceeds to compute embeddings using the specified PLMs (see Step 2).
Step 2: Embedding Computation¶
In this stage, FANTASIA generates numerical representations (embeddings) for each input protein sequence using one or more pretrained protein language models (PLMs). These embeddings serve as the foundation for downstream functional annotation.
Each model is applied independently, and sequences are embedded individually to avoid artifacts caused by padding or batching effects (Configurable, but we found strange behaviours when batching). The embeddings are extracted from the final hidden layer of each model and stored in a shared HDF5 file, which also retains the original sequences for consistency and traceability.
This embedding file becomes the central data structure for the annotation process. It is reused later during similarity search and annotation transfer, and may also be archived or shared for reproducibility.
Internally, this step includes:
Tokenizing each amino acid sequence according to the vocabulary of the corresponding PLM.
Forward-passing the sequences through each model to obtain per-residue and per-sequence representations.
Storing the resulting vectors efficiently in an HDF5 structure, grouped by model and sequence.
This step is executed automatically unless a precomputed embedding file is provided (in which case the pipeline skips directly to the annotation phase).
Step 3: Reference Loading, Filtering and Annotation Transfer¶
In this stage, FANTASIA compares the query embeddings against a curated reference database [6] of experimentally annotated proteins in order to transfer functional annotations (GO terms).
The process begins by loading two reference tables from the internal PostgreSQL database:
Embedding table: Contains high-dimensional vectors previously computed for proteins with experimental GO annotations.
Annotation table: Contains GO terms (excluding electronic inferences) associated with the proteins in the embedding table.
Once these are loaded, FANTASIA applies two optional filters to refine the reference set:
Taxonomy filtering: Entries from specific organisms can be excluded or explicitly included based on NCBI taxonomy IDs. This is useful for benchmarking scenarios where cross-species contamination must be avoided.
Redundancy filtering: To prevent inflated performance due to highly similar sequences, MMseqs2 [2] is used to cluster the query and reference sequences. For each query, any reference entries within the same cluster (i.e., exceeding a given identity and coverage threshold) are excluded from the annotation search.
After filtering, the functional transfer proceeds:
Distance computation: FANTASIA computes the similarity between each query embedding and the remaining reference embeddings, model by model, using a chosen distance metric (e.g., cosine or Euclidean).
Neighbor selection: For each query and model, the top-k most similar entries are selected (typically k = 1 or k = 5). A distance threshold ensures that only close-enough matches are used.
GO term transfer: The GO terms associated with the selected reference proteins are transferred to the query, forming the initial set of functional predictions.
At this point, all predictions are aggregated and stored for post-processing. Each prediction retains metadata including the matched reference, model used, embedding distance, and raw GO term list.
This stage is the core of the FANTASIA pipeline, as it determines the candidate functions for each protein based solely on learned representations rather than sequence alignment.
Step 4: Annotation Post-processing and Output Generation¶
After functional transfer, FANTASIA performs a comprehensive post-processing step to enhance prediction quality, interpretability, and downstream usability.
Reliability Index (RI)¶
FANTASIA assigns a Reliability Index (RI) to each predicted GO term based on its embedding distance to the reference:
Cosine distance:
\[RI = 1 - d_c(q, n_i)\]Euclidean distance:
\[RI = \frac{0.5}{0.5 + d_e(q, n_i)}\]
These scores range from 0 to 1 but are not comparable across metrics. Euclidean-based RI values are model-dependent, while cosine-based RI is better suited for inter-model comparison.
GO Term Prioritization and Filtering¶
FANTASIA retains all predicted GO terms and uses the following metrics to prioritize annotations:
Support count: Number of top hits transferring the same GO term.
RI: Confidence score based on embedding similarity.
For each (accession, go_id, model) triplet, only the prediction with the highest RI is kept in the final table.
Leaf Term Selection and Ontology Collapsing¶
To reduce redundancy and focus on specific functions, FANTASIA collapses ancestor GO terms under their most specific leaf terms using GOATOOLS [7]. It uses the GO ontology graph to identify non-ancestor terms and aggregates broader terms under them:
collapsed_support: Total supporting hits from collapsed ancestors.
n_collapsed_terms: Number of collapsed GO terms.
collapsed_terms: List of absorbed GO terms.
This step ensures annotations are concise and biologically informative.
Pairwise Sequence Alignment¶
For interpretability, FANTASIA includes optional global sequence alignment using Parasail [9], applying:
Needleman-Wunsch algorithm
Substitution matrix: BLOSUM62
Gap penalties: open = 10, extension = 1
The alignment quantifies sequence-level similarity between the query and reference proteins supporting a GO term but does not affect annotation transfer.
Metrics stored include:
identity, similarity, alignment_score
gaps_percentage, alignment_length
length_query, length_reference
TopGO Compatibility¶
FANTASIA exports a TSV file (results_topgo.tsv) compatible with the topGO R package [8], listing comma-separated GO terms per query. This facilitates enrichment analysis in downstream workflows.
Output Files¶
All outputs are stored under a timestamped folder in ~/fantasia/experiments/.
results.csv: Final deduplicated annotations per query
raw_results.csv: All transferred annotations before filtering
results_topgo.tsv: Format for GO enrichment tools
embeddings.h5: Sequence and embedding data used in lookup
These files include both prediction metadata and alignment statistics, allowing advanced filtering and reproducibility.